Sensitive Filtering of Noantigenes¶
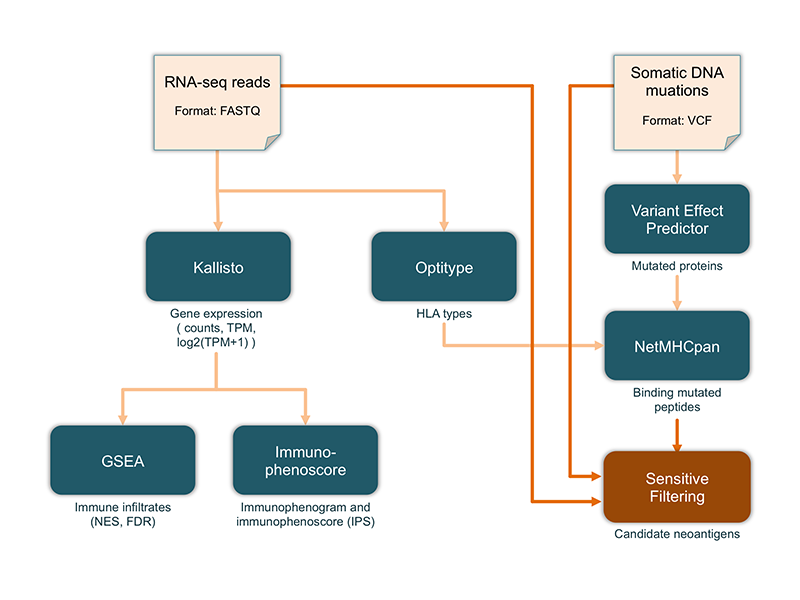
The sensitive filtering module of the TIminer pipeline allows selecting expressed neoantigens considering the allele-specific expression. As shown in the following figure, the sensitive filtering is applied on the mutated binding peptides predicted by NetMHCpan, and considers the list of DNA mutations (VCF format) and the RNA-seq reads (FASTQ format). The filtering is achieved through three main computational steps: (1) sensitive mapping of the RNA-seq reads with HiSat2 [1]; (2) calculation of the RAN-seq read coverage for each mutation with ASEReadCounts function from the Genome Analysis Toolkit (GATK) [2]; and (3) filtering of expressed mutated peptides with a read coverage greater or equal than 5 counts. The sensitive filtering of neoantigens can be activated in the TIminer pipeline launched from command line by specifing in the –sensitivefiltering parameter:
usage: python TIminerPipeline.py [-h] --input INPUT --out OUT [--database DATABASE] [--threadcount THREADCOUNT] [--sensitivefiltering]
Output files¶
The filtered-neoantigens directory contains the files:
- subjectID_neoantigens_sensitiveFiltered.txt: a
filefor each subject containing only the expressed neoantigens; it has the same columns as the files generated by NetMHCpan, plus an additional field reporting the expression read coverage for the mutated allele.
Sensitive filtering functions¶
Single-Subject data analysis
-
TIminer.TIminerAPI.sensitiveFilterNeoantigen(vcf, neoantigenCandidates, fastq1, fastq2=None, filteredneoantigens=None, genomeVersion=38, subject='unkonwn', countThresh=5, pairedEndAlignment='fr', minBaseQuality=20, minMappingQuality=25, threads=2)¶ This function considers a file of candidate neoantigens, and filters them regarding to their mutation specific expression.
Parameters: - vcf (str) – Path to the input file of somatic DNA mutations. The supported format is the VCF format.
- neoantigenCandidates (str) – Path to the
neoantigen input fileas generated by the Netmhcpan, containing binding peptides as possible neoantigen canditates (see the output files section for details). - fastq1 (str) – Path to the first
FASTQ filecontaining the RNA-seq reads. - fastq2 (str) – For paired-end data, path to the second
FASTQ fileof RNA-seq reads (optional). - filteredneoantigens (str) – Path to the
output file(optional, default = mutated-proteins/subject_neoantigens_sensitiveFiltered.txt). - genomeVersion (int) – Version of the human genome used as reference annotation, being 37 for Hg37, or 38 for Hg38 (optional, default = 38).
- subject (str) – Subject ID to be stored in the result file (optional, default = unknown).
- countThresh (int) – Allele count threshold, we consider an allele having five mapped transcripts as expressed (optional, default = 5).
- pairedEndAlignment (str) – The upstream/downstream mate orientations for a valid paired-end alignment against the forward reference strand. E.g., if ‘fr’ is specified and there is a candidate paired-end alignment where mate 1 appears upstream of the reverse complement of mate 2. Other valid options are ‘fr’, ‘rf’, ‘ff’ (optional - just applies for paired-end reads, default = fr).
- minBaseQuality (int) – Minimum base quality (optional, default = 20).
- minMappingQuality (int) – Minimum read mapping quality (optional, default = 25).
- threads (int) – Number of threads to be used (optional, default = 2).
Multiple-Subject data analysis
-
TIminer.TIminerAPI.sensitiveFilterNeoantigenDir(inputInfoFile, outputDir=None, genomeVersion=38, countThresh=5, pairedEndAlignment='fr', minBaseQuality=20, minMappingQuality=25, threads=2)¶ This function considers a file of candidate neoantigens for each subject, and filters them regarding to their mutation specific expression.
Parameters: - inputInfoFile (str) – Path to the
input info file: a tab-delimited file specifying, for each subject: the subject ID, the path to the input RNA-seq files (RNAseq1.fastqandRNAseq2.fastq), the path the noantigen candidates generated by the output of NetMHCpan and the path to the mutation files (mutation1,mutation2, etc.). In case of single-end data, the second RNA-seq file must be simply specified as “None”. One mutation file is mandatory; multiple mutation files can be specified in additional columns. - outputDir (str) – Path to the output directory where the output files will be saved (optional, default = mutated-proteins/).
- genomeVersion (int) – Version of the human genome used as reference annotation, being 37 for Hg37, or 38 for Hg38 (optional, default = 38).
- countThresh (int) – Allele count threshold, we consider an allele having five mapped transcripts as expressed (optional, default = 5).
- pairedEndAlignment (str) – The upstream/downstream mate orientations for a valid paired-end alignment against the forward reference strand. E.g., if ‘fr’ is specified and there is a candidate paired-end alignment where mate 1 appears upstream of the reverse complement of mate 2. Other valid options are ‘fr’, ‘rf’, ‘ff’ (optional - just applies for paired-end reads, default = fr).
- minBaseQuality (int) – Minimum base quality (optional, default = 20).
- minMappingQuality (int) – Minimum read mapping quality (optional, default = 25).
- threads (int) – Number of threads to be used (optional, default = 2).
- inputInfoFile (str) – Path to the